
Discurso del profesor Kenneth S.W. Sing "Los aspectos históricos de la absorción física de gases por sólidos"Con motivo de su investidura como Doctor Honoris Causa en Ciencias por la UNED | ||
|
1. Antecedentes históricos
En 1909 McBain escribió [1] que la fijación de hidrógeno por un carbón parecía ocurrir en dos etapas: un proceso rápido de adsorción era seguido por otro lento de absorción en el interior del sólido. McBain propuso el uso del término sorción para cubrir ambos fenómenos. Incluso en la actualidad, en algunos sistemas es difícil distinguir claramente entre la adsorción en la interfase gas/sólido y la absorción en la estructura del sólido. En ese caso es conveniente usar el término sorción, junto con el de sorbente, sorbato y sortivo [2].
Los primeros trabajos sobre adsorción física (fisisorción) han sido reservados por Brunauer [4], Deitz [5] y Forrester y Giles [6]. 2. El concepto de monocapa
| ||
|
|
||||||||||||||||||
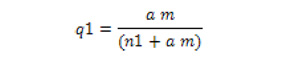
|
donde q1 es la fracción de superficie cubierta por las moléculas adsorbidas, m es el número moléculas colisionantes por centímetro cuadrado de superficie y por segundo, a es un coeficiente de condensación (a<1) y n1 es la velocidad de evaporación a partir de una superficie completamente cubierta. De acuerdo con la teoría de la colisión, m es proporcional a la presión del gas, p, y por tanto, la expresión (1) la podemos transformar en la conocida ecuación de Langmur (8): | ||
|
|
||||||||||||||||||
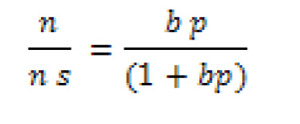
|
donde ns y b son constantes empíricas. Por supuesto que si el modelo de Langmuir se cumple, ns, debe corresponderse con el límite de cubrimiento de q=1 (i.e. la capacidad de la monocapa).
| ||
|
3. Adsorción en multicapa
En condiciones de equilibrio, las velocidades de condensación y evaporación se igualan para cada capa adsorbida y, se asumió, que por un lado, las constantes de velocidad eran las mismas para todas las capas después de la primera y que, por otro, la energía de adsorción para las capas superiores era igual al valor de la energía de condensación. La suma de las cantidades adsorbidas en todas las capas dio una ecuación bastante simple para la isoterma de adsorción. | ||
|
|
||||||||||||||||||

donde nm es la capacidad de la monocapa y C una constante. De acuerdo con la teoría de BET, C está relacionada exponencialmente con la energía de adsorción de la primera capa.
4. Condensación Capilar
| ||
|
|
||||||||||||||||||
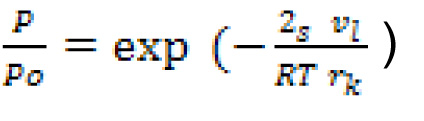
|
donde s es la tensión superficial y v, el volumen molar del líquido adsorbible. La ecuación (4), que es una forma modificada de la relación originalmente propuesta por Lord Kelvin (W. Thomson) en 1871, es, universalmente conocida, como ecuación de Kelvin. El rango de aplicabílidad de la ecuación de Kelvin ha sido discutido durante más de 75 años. Esta ecuación es ampliamente aplicada actualmente para el análisis del tamaño de los poros, pero algunas de sus limitaciones quedan por resolver.
Madrid, junio 1996 | ||
|
6. Bibliografía/References 1. McBain, J.W.; Phil. Mag., 18, 916 (1909). 2. Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L, Pierotti, R.A., Rouque-rol, J. and Siemieniewska, T.; Puré Appl. Chem., 57, 603-619 (1985). 3. Lennard-Jones, J.E.; The Adsorption of Gases by Solids, Farad. Soc, 333 -359(1932). 4. Brunauer, S.; uThe Adsorption of Gases and Vapours", Oxford University Press (1945). 5. Deitz, V.R.; "Bibliography of Solid Adsorbents", National Bureau of Standards Washington (1944). 6. Forrester, S.D. and Giles, C.H.; Chem. & Ind., 831-839 (1971). 7. Langmuir, I.; J. Amer. Chem. Soc, 38, 2221-2295 (1916). 8. Langmuir, I.; J. Amer. Chem. Soc, 40, 1361-1403 (1918). 9. Brunauer, S. and Emmett, P.H.; J. Amer. Chem. Soc, 59, 2682 (1937).
10. Brunauer, S., Emmett, P.H. and Teller, E.; J. Amer. Chem. Soc, 60, 309-319 (1938). 11. Brunauer, S. and Emmett, P.H.; J. Amer. Chem. Soc, 57,1754 (1935). 12. Brunauer, S., Deming, L.S., Deming, W.E. and Teller, E.; J. Amer. Chem. Soc, 57, 1754(1940). 13 Young, D.M. and Crowell, A.D.; "Physical Adsorption of Gases", Butterworths, London (1962). 14. Zsigmondy, R.; Z anorg. Chem., 71, 356 (1911). 15. Foster, A.G.; Trans. Farad. Soc, 28, 645 (1932). 16. McBain, J.W.; The Adsorption of Gases by Solids, Farad. Soc, 408-409 (1932). 17. Barrer, R.M.; "Zeolites and Clay Minerals as Sorbents and Molecular Sieves", Academic Press, London (1978). 18. Dubinin, M.M.; J. Colloid Ihteríace Sci., 23, 487-499 (1967). 19. Kiselev, A.V.; Discuss. Farad. Soc, .40, 205-218 (1965). 20. Sing, K.S.W.; "Surface Área DeterminatiorT, 25-34, Butterwoths, London (1970). 21. Gregg, S.J. and Sing, K.S.W.; "Adsorption, Surface Área and Porosity, Aca-demic Press, London (1982). 22. Sing, K.S.W.; Colloids & Surfaces, 38,113-124 (1989). 23. Rodriguez-Reinoso, F.; Puré Appl. Chem., 61,1859-1866 (1989). 24. Everett, D.H. and Powl, J.C.; J. Chem. Soc, Farad. Trans. I, 72, 619-636 (1976). 25. Sing, K.S.W.; J. Porous Mat., 2, 5-8 (1995). | ||